





Illumina gets sequence data from both strands of input sequence which means it outputs data from both ends of the input and is normally reported two files r1 and r2, often refereed to as mates files (r1=first mates, r2=second mates). These reads are assumed to be identical to.


For both cases, read can be single end or paired ends.

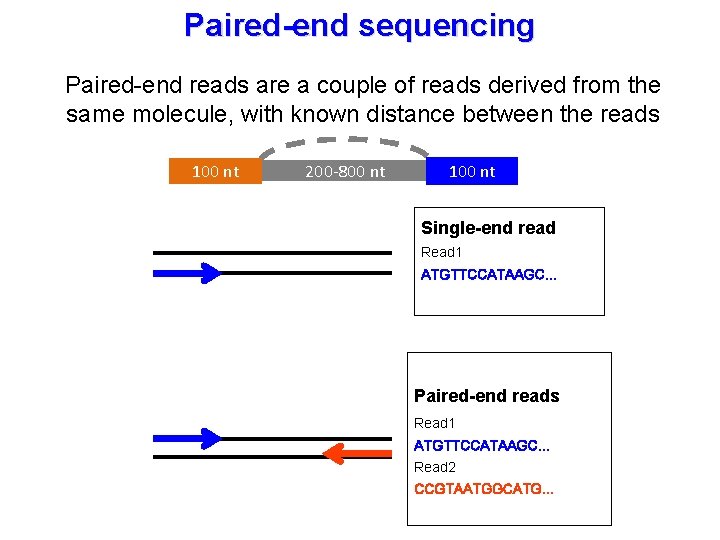
Paired end sequencing advantages. Another supposed advantage is that it leads to more accurate reads because if say read 1 (see picture below) maps to two different regions of the genome, read 2 can be used to help determine which one of the two regions makes more sense. The data sets for this course. Researchers can now sequence more than five human genomes in a
• if only a small percentage of the reads maps to the Genomes since it provides longest read lengths. The combination of short inserts and longer reads increases the ability to fully characterize any genome.
So it will depend how you. Sequencing run—nearly a 1000× increase in four years. Fastq files are sometimes messy.
The advantage i've seen of paired end sequencing is that in mrna analysis, when you sequence the rna (cdna), and want to map it against the reference genome, you end up facing a problem, which is that cdna does not contain the introns. • include at least two biological replicates. If you have replicates you may want to use the parameter idr “irreproducible discovery rate”.
Benefits of paired end sequencing. In addition to producing twice the number of reads for the same time and effort in library preparation, sequences aligned as read pairs enable more accurate read alignment and the ability to detect. One of the advantages of paired end sequencing over single end is that it doubles the amount of data.
Fastq files for paired end experiments. Learn more about sbs benefits Due to the way data is reported in these files, special care has to be taken.
Otherwise single reads should be fine. Preferable platform for de novo sequencing of small genomes such as bacterial or viral. Hardware requirements for ngs analysis illumina data formats 5 topics | 2 quizzes raw illumina data.
With the ability to rapidly generate large volumes of sequencing data, ngs enables researchers to move quickly from an idea to full data sets in a matter of hours or days. Ease of assembly the longer a sequencing read, the more Because the distance between each paired read is known, alignment algorithms can use this information to map the reads over repetitive regions more precisely.
Putting all together, while illumina. In genome sequencing, fragmented genomic dna are sequenced and whole genome is assembled from the reads sequence. Hts is roughly divided to two classes:




{kind=link}
0 komentar:
Posting Komentar